
Tue, Jun 25, 2024

Volume 16, Issue 6 (Special issue (Nov-Dec) 2022)
mljgoums 2022, 16(6): 26-34 |
Back to browse issues page

Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Prajapat R, Jain S. Effective Binding Affinity of Inhibitor N-(3-(Carbamoylamino) Phenyl) Acetamide against the SARS-CoV-2 NSP13 Helicase. mljgoums 2022; 16 (6) :26-34
URL: http://mlj.goums.ac.ir/article-1-1475-en.html
URL: http://mlj.goums.ac.ir/article-1-1475-en.html



1- Department of Biochemistry, Pacific Institute of Medical Sciences, Sai Tirupati University, Udaipur, Rajasthan, India , rajneesh030041@gmail.com
2- Department of Biochemistry, Pacific Institute of Medical Sciences, Sai Tirupati University, Udaipur, Rajasthan, India
2- Department of Biochemistry, Pacific Institute of Medical Sciences, Sai Tirupati University, Udaipur, Rajasthan, India
Full-Text [PDF 750 kb]
(269 Downloads)
| Abstract (HTML) (1463 Views)
Full-Text: (409 Views)
INTRODUCTION
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the causative agent of the ongoing coronavirus disease 2019 (COVID-19) pandemic (1, 2). The novel coronavirus was first reported from Wuhan, Hubei Province of China in late 2019 (3-5). The virus has spread worldwide within a year (6-8). The SARS-CoV-2 is a member of the family Coronaviridae (9). Coronaviruses are enveloped, single-stranded RNA viruses with genome sizes ranging from 26 to 32 kb (10-12) and at least 6 (14 in case of SARS-CoV-2) open reading frames (13, 14). The first genome sequence of SARS-CoV-2 deposited in the GenBank was reported from Wuhan-Hu-1 (MN908947) as a ~30 kb isolate (15), which was used for sequence-related analyses in this article.
The ongoing outbreak of SARS-CoV-2 has caused tremendous economic and human losses around the world, while there is still no approved drug available for this infection. The current COVID-19 pandemic has prompted worldwide efforts for the rapid identification and development of vaccines and effective antiviral treatments (16, 17). Most previous studies on anti-coronavirus targets have been focused on the main protease, spike protein (S), RNA-dependent RNA polymerase (RdRp, NSP12), NTPase/ NSP13 helicase, and papain-like protease (PLpro, part of NSP3) (18-20). The SARS-CoV-2 NSP13 possesses NTPase and RNA helicase activities and is conserved (21). The NTPase and helicase activities of SARS-CoV-2 NSP13 may play an important role in SARS-CoV-2 replication and could serve as a target for antivirals (20). In addition, NSP13 ZBD conformations indicate the role of induced-fit flexibility at the ligand binding site (40). Drug hits have significant activity in inhibiting recombinant SARS-CoV-2 helicase in its apo- and ATP/RNA-bound conformations (46). The present study was carried out to investigate the potential application of inhibitors against SARS-CoV-2 NSP13 helicase.
MATERIALS AND METHODS
Sequence retrieval and homology modeling
The amino acid sequence of SARS-CoV-2 NSP13 helicase (Accession number: YP_009725308) was retrieved from NCBI (601 aa) in FASTA format (22). PSI-BLAST was used for alignment against the Protein Databank (PDB) for a suitable template search with query sequence (23). The template with query sequence (PDB ID: 5WWP_A) was selected having a 72.20% identity score and 0.00 E-value. The SWISS-MODEL (https://swissmodel.expasy.org) structure assessment tool was used for homology modeling and visual analysis of the crystal structure of the protein (24).
PROCHECK server was used for validation of the structure model (25, 26), and its results suggested the reliability of the model (27). The overall G-factor and residue positions in φ-ψ plot regions analysis were used for the selection of a suitable model. The quality of the 3D model was verified by ERRAT, Verify3D, and ANOLEA. ERRAT plot confirmed the overall quality of the model (28).
Protein stability was analyzed by using SWISS-MODEL QMEAN (version 4.2.0) (29) and ProSA Z-score. The protein structure analysis (ProSA) calculated an overall quality score for the predicted structure (30).
Molecular docking
Molecular docking techniques dock small molecules into the protein binding site (31). The MCULE-1-Click docking (https://mcule.com) and InterEvDock 2.0 (https://bioserv.rpbs.univ-paris-diderot.fr/) programs were used for docking calculations (32). 1-Click docking is an online server for drug discovery platforms (33).
The study protocol received approval from the Ethics Committee of the Pacific Institute of Medical Sciences, Sai Tirupati University, Udaipur, Rajasthan, India (Registration no: STU/2019/5006).
RESULTS
Protein model building
The alignment between the target and template was performed using PSI-BLAST (22). The 3D ribbon model of SARS-CoV-2 NSP13 helicase was generated using SWISS-MODEL (https://swissmodel.expasy.org) structure assessment tool (Figure 1).
.PNG)
.PNG)
.PNG)
Table 1- Evaluation of the protein model by PROCHECK VERIFY3D, and ANOLEA
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the causative agent of the ongoing coronavirus disease 2019 (COVID-19) pandemic (1, 2). The novel coronavirus was first reported from Wuhan, Hubei Province of China in late 2019 (3-5). The virus has spread worldwide within a year (6-8). The SARS-CoV-2 is a member of the family Coronaviridae (9). Coronaviruses are enveloped, single-stranded RNA viruses with genome sizes ranging from 26 to 32 kb (10-12) and at least 6 (14 in case of SARS-CoV-2) open reading frames (13, 14). The first genome sequence of SARS-CoV-2 deposited in the GenBank was reported from Wuhan-Hu-1 (MN908947) as a ~30 kb isolate (15), which was used for sequence-related analyses in this article.
The ongoing outbreak of SARS-CoV-2 has caused tremendous economic and human losses around the world, while there is still no approved drug available for this infection. The current COVID-19 pandemic has prompted worldwide efforts for the rapid identification and development of vaccines and effective antiviral treatments (16, 17). Most previous studies on anti-coronavirus targets have been focused on the main protease, spike protein (S), RNA-dependent RNA polymerase (RdRp, NSP12), NTPase/ NSP13 helicase, and papain-like protease (PLpro, part of NSP3) (18-20). The SARS-CoV-2 NSP13 possesses NTPase and RNA helicase activities and is conserved (21). The NTPase and helicase activities of SARS-CoV-2 NSP13 may play an important role in SARS-CoV-2 replication and could serve as a target for antivirals (20). In addition, NSP13 ZBD conformations indicate the role of induced-fit flexibility at the ligand binding site (40). Drug hits have significant activity in inhibiting recombinant SARS-CoV-2 helicase in its apo- and ATP/RNA-bound conformations (46). The present study was carried out to investigate the potential application of inhibitors against SARS-CoV-2 NSP13 helicase.
MATERIALS AND METHODS
Sequence retrieval and homology modeling
The amino acid sequence of SARS-CoV-2 NSP13 helicase (Accession number: YP_009725308) was retrieved from NCBI (601 aa) in FASTA format (22). PSI-BLAST was used for alignment against the Protein Databank (PDB) for a suitable template search with query sequence (23). The template with query sequence (PDB ID: 5WWP_A) was selected having a 72.20% identity score and 0.00 E-value. The SWISS-MODEL (https://swissmodel.expasy.org) structure assessment tool was used for homology modeling and visual analysis of the crystal structure of the protein (24).
PROCHECK server was used for validation of the structure model (25, 26), and its results suggested the reliability of the model (27). The overall G-factor and residue positions in φ-ψ plot regions analysis were used for the selection of a suitable model. The quality of the 3D model was verified by ERRAT, Verify3D, and ANOLEA. ERRAT plot confirmed the overall quality of the model (28).
Protein stability was analyzed by using SWISS-MODEL QMEAN (version 4.2.0) (29) and ProSA Z-score. The protein structure analysis (ProSA) calculated an overall quality score for the predicted structure (30).
Molecular docking
Molecular docking techniques dock small molecules into the protein binding site (31). The MCULE-1-Click docking (https://mcule.com) and InterEvDock 2.0 (https://bioserv.rpbs.univ-paris-diderot.fr/) programs were used for docking calculations (32). 1-Click docking is an online server for drug discovery platforms (33).
The study protocol received approval from the Ethics Committee of the Pacific Institute of Medical Sciences, Sai Tirupati University, Udaipur, Rajasthan, India (Registration no: STU/2019/5006).
RESULTS
Protein model building
The alignment between the target and template was performed using PSI-BLAST (22). The 3D ribbon model of SARS-CoV-2 NSP13 helicase was generated using SWISS-MODEL (https://swissmodel.expasy.org) structure assessment tool (Figure 1).
Table 1- Evaluation of the protein model by PROCHECK VERIFY3D, and ANOLEA
| Template | PROCHECK | Verify-3D | ANOLEA | |||
NSP13 |
Core | Allowed | Generously outer | Disallowed | 3D-ID Score |
Z-Score |
| 90.9% | 8.5% | 0.4 % | 0.2 % | 95.9 | -0.35 | |
Molecular dockingThe binding pockets of SARS-CoV-2 NSP13 helicase are not reported yet. Thus, the in silico approaches were utilized for the prediction of binding pockets. MCULE-1-Click docking (https://mcule.com) and InterEvDock-2.0 server were employed to explore the binding of ligands to the respective protein. The top five docking models of binding pockets of SARS-CoV-2 NSP13 helicase were identified and ranked based on the energy. More negative docking scores indicated higher binding affinity (Table 2). The summary table contains two rows: the ranks and docking energy scores from the input structures. Model 1 has high accuracy with an interface docking score of -5.940 kcal/mol from the crystal structure (Table 2).
Table 2- Summary of the top five models. Row 1 (ranks of the models), Row 2 (docking energy scores - kcal/mol)
Table 2- Summary of the top five models. Row 1 (ranks of the models), Row 2 (docking energy scores - kcal/mol)
| Rank | 1 | 2 | 3 | 4 | 5 |
| Docking Score | -5.940 | -5.920 | -5.790 | -5.440 | -5.430 |
| Rank | Energy (Kcal/mol) | Center | ||||
| X-axis | Y-axis | Z-axis | ||||
| 1 | -5.940 | 46.4820 | 13.1600 | 25.1190 | ||
| 2 | -5.920 | 45.8500 | 14.4180 | 25.0310 | ||
| 3 | -5.790 | 45.4730 | 15.1320 | 26.1920 | ||
| 4 | -5.440 | 46.7390 | 12.5780 | 26.3880 | ||
| 5 | -5.430 | 46.3440 | 13.2850 | 27.5460 | ||
Model reputation
The SARS-CoV-2 NSP13 helicase model corresponding to probability confirmation with 90.9% residue of the core section, 8.5% of the allowed section, and 0.4 % residue of the outer section in the φ-ψ plot (34) (Figure 2a, b). The above results indicate the reliability of protein models (Table 1) (35, 36).
The verify-3D illustrates the compatibility of an atomic model (3D) with its amino acid sequence (1D) by assigning a structural class based on its location and environment (alpha, beta, loop, polar, and nonpolar) (37). ANOLEA recorded non-local energy of the helicase was -6028 E/kT units, with a Z-score of -0.35. These scores indicate the good quality of the model (range 0.16 to -0.02) (Table 1).
ERRAT analyses the statistics of non-bonded interactions between different atom types and plots the value of the error function versus position, which is calculated by comparison with statistics from highly refined structures (38). ERRAT overall quality factor of the model was 93.5829, with an average probability value of 5.05729 (Figure 3).
Validation of the model
ProSA was used to determine the potential errors in the 3D model of SARS-CoV-2 NSP13 helicase. The archived ProSA Z-score of -9.17 indicates two aspects: overall model quality and energy deviation (Figure 4).
.PNG)
.PNG)
The QMEAN4 value of -0.99 was observed for the NSP13 helicase, which is very close to 0 and therefore an acceptable value (44). Assessed validity of the model predictable among 0 and 1, which could be concluded from the density plot locus set for QMEAN score (Figure 5). Figure 5 illustrates the QMEAN scores for the biological unit reference set, which were used as a tool for oligomeric protein assessment.
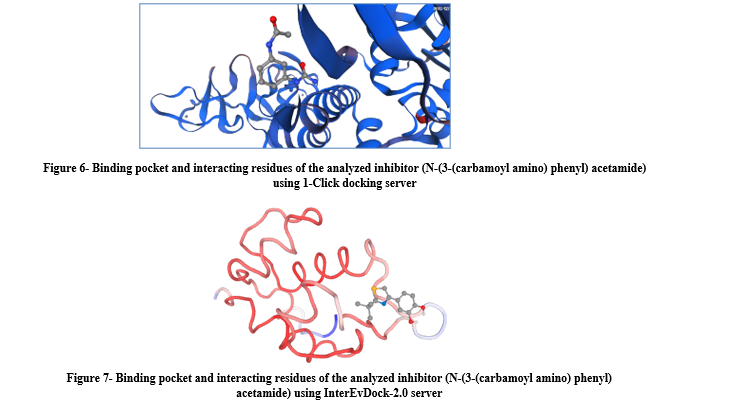
The binding pocket and interacting residues of the selected inhibitor (N-(3-(carbamoyl amino) phenyl) acetamide) was analyzed in 3D using both servers (Figures 6 and 7). The binding residues of the cavities were explored for the fruitful binding of novel ligands. The energy range of predicted cavities also indicated the efficacy of pockets. The mutational study of binding residues suggested that these residues could be used as a clinical prospectus for the effective treatment of COVID-19. The predicted binding residues lead to the drug designing of lead compounds against SARS-CoV-2 NSP13 helicase.
Table 4- Properties and binding residues of the inhibitor N-(3-(carbamoyl amino) phenyl) acetamide (PubChem CID: 828139)
It was observed that the inhibitor binds at the binding residues between Lys-146 and Ser-229 (Table 4). The predicted structural and docking model described in this study may be further used for finding interactions with other SARS-CoV-2 enzymes to identify new anti-coronavirus targets.
DISCUSSION
In silico structural analysis revealed two ligand binding pockets on NSP13 that are the most conserved sites in the entire SARS-CoV-2 proteome (39). NSP13 ZBD conformations show the role of induced-fit flexibility in the ligand binding site (40). The SARS-CoV-2 NSP13 helicase model corresponds to probability confirmation with 90.9% residue of the core section that specifies the accuracy of the predicted model. In the verify-3D graph, 95.95% of the residues have averaged a 3D-1D score ≥0.2 which illustrates the results of good structures. ANOLEA recorded non-local energy of the helicase was -6028 E/kT units, with a Z-score of -0.35. These scores indicate the good quality of the model. ERRAT overall quality factor of the model was 93.5829 with an average probability value of 5.05729, which indicates a much more reliable and satisfactory model. The archived ProSA Z-score score of -9.17 also indicates two aspects: overall model quality and energy deviation. The values of the Z-score indicate fewer erroneous structures (41, 42). Reliability of the projected model based on the scoring function of QMEAN that stated as ‘Z-score’ (43).
Assessed validity of model predictable among 0 and 1, which could be concluded from the density plot locus set for QMEAN score. The QMEAN value comparison with the non-redundant protein collection revealed a different set of Z-values for different parameters. The diversion of the total energy of NSP13 helicase was measured by using Z-score (45).
Drug hits have a significant role in inhibiting recombinant SARS-CoV-2 helicase in its apo- and ATP/RNA-bound conformations (46). The potential compounds 26-deoxyactein and 25-O-anhydrocimigenol-3-O-beta-d-xylopyranoside pose strong interactions and sustained close contact with NSP13 (47). The inhibitor N-(3-(carbamoylamino) phenyl) acetamide exhibited effective binding affinity against NSP13 helicase. The docking results revealed the Lys-146, Leu-147, Ile-151, Tyr-185, Lys-195, Tyr-224, Val-226, Leu-227, and Ser-229 residues exhibit good binding interactions with inhibitor ligand N-(3-(carbamoyl amino) phenyl) acetamide. The predicted binding residues serve as the base for drug designing of lead compounds against SARS-CoV-2 NSP13 helicase, which is thought to play key roles during the replication of viral RNAs.
CONCLUSION
The results of this study establish N-(3-(carbamoylamino) phenyl) acetamide as a valuable lead molecule with great potential for SARS-CoV-2 NSP13 helicase inhibition.
ACKNOWLEDGEMENTS
The authors are grateful to Dr. Indrajeet Singhvi (Vice Chancellor of the Sai Tirupati University, Udaipur, Rajasthan, India) for his precious support and guidance during Ph.D. coursework. The biochemistry and bioinformatics research group members are also acknowledged for technical support.
DECLARATIONS
FUNDING
The authors did not receive any financial support for the research and publication of this article.
Ethics approvals and consent to participate
The study protocol received approval from the Ethics Committee of Pacific Institute of Medical Sciences, Sai Tirupati University, Udaipur, Rajasthan, India (Registration no: STU/2019/5006).
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest regarding the publication of this article.
The SARS-CoV-2 NSP13 helicase model corresponding to probability confirmation with 90.9% residue of the core section, 8.5% of the allowed section, and 0.4 % residue of the outer section in the φ-ψ plot (34) (Figure 2a, b). The above results indicate the reliability of protein models (Table 1) (35, 36).
The verify-3D illustrates the compatibility of an atomic model (3D) with its amino acid sequence (1D) by assigning a structural class based on its location and environment (alpha, beta, loop, polar, and nonpolar) (37). ANOLEA recorded non-local energy of the helicase was -6028 E/kT units, with a Z-score of -0.35. These scores indicate the good quality of the model (range 0.16 to -0.02) (Table 1).
ERRAT analyses the statistics of non-bonded interactions between different atom types and plots the value of the error function versus position, which is calculated by comparison with statistics from highly refined structures (38). ERRAT overall quality factor of the model was 93.5829, with an average probability value of 5.05729 (Figure 3).
Validation of the model
ProSA was used to determine the potential errors in the 3D model of SARS-CoV-2 NSP13 helicase. The archived ProSA Z-score of -9.17 indicates two aspects: overall model quality and energy deviation (Figure 4).
The QMEAN4 value of -0.99 was observed for the NSP13 helicase, which is very close to 0 and therefore an acceptable value (44). Assessed validity of the model predictable among 0 and 1, which could be concluded from the density plot locus set for QMEAN score (Figure 5). Figure 5 illustrates the QMEAN scores for the biological unit reference set, which were used as a tool for oligomeric protein assessment.
The binding pocket and interacting residues of the selected inhibitor (N-(3-(carbamoyl amino) phenyl) acetamide) was analyzed in 3D using both servers (Figures 6 and 7). The binding residues of the cavities were explored for the fruitful binding of novel ligands. The energy range of predicted cavities also indicated the efficacy of pockets. The mutational study of binding residues suggested that these residues could be used as a clinical prospectus for the effective treatment of COVID-19. The predicted binding residues lead to the drug designing of lead compounds against SARS-CoV-2 NSP13 helicase.
Table 4- Properties and binding residues of the inhibitor N-(3-(carbamoyl amino) phenyl) acetamide (PubChem CID: 828139)
| Ligand Properties | C9 H11 N3 O2 |
| Molecular weight (g/mol) | 193.203 |
| Component type | Non-polymer |
| Hydrogen bond donor count | 03 |
| Hydrogen bond donor count | 02 |
| Rotatable bonds count | 02 |
| Topological polar surface area | 84.2 Ų |
| Heavy atom count | 14 |
| Formal charge | 0 |
| Interacting residues | Lys-146, Leu-147, Ile-151, Tyr-185, Lys-195, Tyr-224, Val-226, Leu-227, Ser-229 |
It was observed that the inhibitor binds at the binding residues between Lys-146 and Ser-229 (Table 4). The predicted structural and docking model described in this study may be further used for finding interactions with other SARS-CoV-2 enzymes to identify new anti-coronavirus targets.
DISCUSSION
In silico structural analysis revealed two ligand binding pockets on NSP13 that are the most conserved sites in the entire SARS-CoV-2 proteome (39). NSP13 ZBD conformations show the role of induced-fit flexibility in the ligand binding site (40). The SARS-CoV-2 NSP13 helicase model corresponds to probability confirmation with 90.9% residue of the core section that specifies the accuracy of the predicted model. In the verify-3D graph, 95.95% of the residues have averaged a 3D-1D score ≥0.2 which illustrates the results of good structures. ANOLEA recorded non-local energy of the helicase was -6028 E/kT units, with a Z-score of -0.35. These scores indicate the good quality of the model. ERRAT overall quality factor of the model was 93.5829 with an average probability value of 5.05729, which indicates a much more reliable and satisfactory model. The archived ProSA Z-score score of -9.17 also indicates two aspects: overall model quality and energy deviation. The values of the Z-score indicate fewer erroneous structures (41, 42). Reliability of the projected model based on the scoring function of QMEAN that stated as ‘Z-score’ (43).
Assessed validity of model predictable among 0 and 1, which could be concluded from the density plot locus set for QMEAN score. The QMEAN value comparison with the non-redundant protein collection revealed a different set of Z-values for different parameters. The diversion of the total energy of NSP13 helicase was measured by using Z-score (45).
Drug hits have a significant role in inhibiting recombinant SARS-CoV-2 helicase in its apo- and ATP/RNA-bound conformations (46). The potential compounds 26-deoxyactein and 25-O-anhydrocimigenol-3-O-beta-d-xylopyranoside pose strong interactions and sustained close contact with NSP13 (47). The inhibitor N-(3-(carbamoylamino) phenyl) acetamide exhibited effective binding affinity against NSP13 helicase. The docking results revealed the Lys-146, Leu-147, Ile-151, Tyr-185, Lys-195, Tyr-224, Val-226, Leu-227, and Ser-229 residues exhibit good binding interactions with inhibitor ligand N-(3-(carbamoyl amino) phenyl) acetamide. The predicted binding residues serve as the base for drug designing of lead compounds against SARS-CoV-2 NSP13 helicase, which is thought to play key roles during the replication of viral RNAs.
CONCLUSION
The results of this study establish N-(3-(carbamoylamino) phenyl) acetamide as a valuable lead molecule with great potential for SARS-CoV-2 NSP13 helicase inhibition.
ACKNOWLEDGEMENTS
The authors are grateful to Dr. Indrajeet Singhvi (Vice Chancellor of the Sai Tirupati University, Udaipur, Rajasthan, India) for his precious support and guidance during Ph.D. coursework. The biochemistry and bioinformatics research group members are also acknowledged for technical support.
DECLARATIONS
FUNDING
The authors did not receive any financial support for the research and publication of this article.
Ethics approvals and consent to participate
The study protocol received approval from the Ethics Committee of Pacific Institute of Medical Sciences, Sai Tirupati University, Udaipur, Rajasthan, India (Registration no: STU/2019/5006).
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest regarding the publication of this article.
Research Article: Research Article |
Subject:
Biochemistry
Received: 2022/01/17 | Accepted: 2022/12/19 | Published: 2022/11/25 | ePublished: 2022/11/25
Received: 2022/01/17 | Accepted: 2022/12/19 | Published: 2022/11/25 | ePublished: 2022/11/25
References
1. Ullrich S, Nitsche C. The SARS-CoV-2 main protease as drug target. Bioorg Med Chem Lett. 2020; 30(17): 127377 [View at Publisher] [DOI:10.1016/j.bmcl.2020.127377] [PubMed] [Google Scholar]
2. Dong E, Du H, Gardner L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect Dis. 2020; 20(5): 533-534. [View at Publisher] [DOI:10.1016/S1473-3099(20)30120-1] [PubMed] [Google Scholar]
3. Gupta B, Kalhan S, Shukla S, Bahadur S, Singh G, Pathak R. Evaluating Association between ABO Blood Groups and COVID 19. mljgoums. 2021; 15 (6) :1-7. [View at Publisher] [Google Scholar]
4. Wu F, Zhao S, B. Yu, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020; 579 (7798): 265-269. [View at Publisher] [DOI:10.1038/s41586-020-2008-3] [PubMed] [Google Scholar]
5. Priya R, Andurkar SP, Dixit JV. Determinants of outcome in covid-19 cases: a cross-sectional analytical study. Al Ameen Journal of Medical Sci. 2021; 14 (1): 39-42. [View at Publisher] [Google Scholar]
6. Chan JFW, Yuan S, Kok KH, et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet. 2020; 395 (10223): 514-523. [View at Publisher] [DOI:10.1016/S0140-6736(20)30154-9] [PubMed] [Google Scholar]
7. Yang Y, Peng F, Wang R, et al. The deadly coronaviruses: the 2003 SARS pandemic and the 2020 novel coronavirus epidemic in China. J Autoimmun. 2020; 109: 102434. [View at Publisher] [DOI:10.1016/j.jaut.2020.102434] [PubMed] [Google Scholar]
8. Adake P, Acharya A, Halemani S, Petimani M. Clinical Features of COVID-19 Patients with Preexisting Hypothyroidism: A Retrospective Study. mljgoums. 2022; 16 (1) :9-12. [View at Publisher] [Google Scholar]
9. Song Z, Xu Y, Bao L, Zhang L, Yu P, Qu Y, Zhu H, Zhao W, Han Y, Qin C. From SARS to MERS, Thrusting Coronaviruses into the Spotlight. Viruses. 2019 Jan 14;11(1):59. [View at Publisher] [DOI:10.3390/v11010059] [PubMed] [Google Scholar]
10. Song Z, Xu Y, Bao L. From SARS to MERS: thrusting coronaviruses into the spotlight. Viruses. 2019; 11(1):59. doi: 10.3390/v11010059. [DOI:10.3390/v11010059]
11. Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003; 300(5626):1763-1767. [View at Publisher] [DOI:10.1126/science.1085658] [PubMed] [Google Scholar]
12. Arun MR, Sheeba MR, Rishma FSF. Historical Analysis and Scientific Overview of Coronaviruses. Al Ameen Journal of Medical Sci. 2020; 13 (03): 141-148. [View at Publisher] [Google Scholar]
13. Chen Y, Liu Q, Guo D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J Med Virol. 2020; 92(4):418-423. [View at Publisher] [DOI:10.1002/jmv.25681] [PubMed] [Google Scholar]
14. Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020; 583(7816): 459-468. [View at Publisher] [DOI:10.1038/s41586-020-2286-9] [PubMed] [Google Scholar]
15. Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265-269. [View at Publisher] [DOI:10.1038/s41586-020-2008-3] [PubMed] [Google Scholar]
16. Zhang C, Huang S, Zheng F, Dai Y. Controversial treatments: an updated understanding of the coronavirus disease 2019. J Med Virol. 2020. [View at Publisher] [DOI:10.1002/jmv.25788] [PubMed] [Google Scholar]
17. Lurie N, Saville M, Hatchett R, Halton J. Developing COVID-19 vaccines at pandemic speed. N Engl J Med. 2020; 382(21):1969-1973. [View at Publisher] [DOI:10.1056/NEJMp2005630] [PubMed] [Google Scholar]
18. Ullrich S, Nitsche C. The SARS-CoV-2 main protease as drug target. Bioorg Med Chem Lett. 2020; 30(17):127377. [View at Publisher] [DOI:10.1016/j.bmcl.2020.127377] [PubMed] [Google Scholar]
19. Wu C, Liu Y, Yang Y. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm Sin. 2020; 10(5):766-788. [View at Publisher] [DOI:10.1016/j.apsb.2020.02.008] [PubMed] [Google Scholar]
20. Shu T, Huang M, Wu D. et al. SARS-Coronavirus-2 Nsp13 Possesses NTPase and RNA Helicase Activities That Can Be Inhibited by Bismuth Salts. Virol Sin. 2020; 35: 321-329. [View at Publisher] [DOI:10.1007/s12250-020-00242-1] [PubMed] [Google Scholar]
21. Habtemariam S, Nabavi SF, Banach M, Berindan-Neagoe I, Sarkar K, Sil PC, et al. Should We Try SARS-CoV-2 Helicase Inhibitors for COVID-19 Therapy?. Arch Med Res. 2020; 51(7):733-735. [View at Publisher] [DOI:10.1016/j.arcmed.2020.05.024] [PubMed] [Google Scholar]
22. Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, et al. The sequence of the human genome. Science. 2001; 291: 1304-1351. [View at Publisher] [DOI:10.1126/science.1058040] [PubMed] [Google Scholar]
23. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990; 215: 403-410. [View at Publisher] [DOI:10.1016/S0022-2836(05)80360-2]
24. Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, Lepore R, Schwede T. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018; 46: W296-W303. [View at Publisher] [DOI:10.1093/nar/gky427] [PubMed] [Google Scholar]
25. Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK - a program to check the stereochemical quality of protein structures. J. App. Cryst. 1993; 26: 283-291. [View at Publisher] [DOI:10.1107/S0021889892009944]
26. Vriend G. WHAT IF: A molecular modeling and drug design program. J Mol Graphics. 1990; 8: 52-56. [View at Publisher] [DOI:10.1016/0263-7855(90)80070-V] [PubMed] [Google Scholar]
27. Sehgal SA, Tahir RA, Shafique S, Hassan M, Rashid S. Molecular modeling and docking analysis of CYP1A1 associated with head and neck cancer to explore its binding regions. J Theoret Comput Sci. 2014; 1 (3):1-6. [DOI:10.4172/2376-130X.1000112] [Google Scholar]
28. Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 1993; 2: 1511-1519. [View at Publisher] [DOI:10.1002/pro.5560020916] [PubMed] [Google Scholar]
29. Benkert P, Kunzli M, Schwede T. QMEAN server for protein model quality estimation. Nucleic Acids Res 2009; 37: W510-W514. [View at Publisher] [DOI:10.1093/nar/gkp322] [Google Scholar]
30. Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007; (35) 2: W407-W410. [View at Publisher] [DOI:10.1093/nar/gkm290] [PubMed] [Google Scholar]
31. Prajapat R, Marwal A, Gaur RK. Recognition of errors in the refinement and validation of three-dimensional structures of AC1 proteins of begomovirus strains by using ProSA-web. J Viruses. 2014; 6. [View at Publisher] [DOI:10.1155/2014/752656] [Google Scholar]
32. Quignot C, Rey J, Yu J, Tufféry P, Guérois R, Andreani J. InterEvDock2: an expanded server for protein docking using evolutionary and biological information from homology models and multimeric inputs. Nucleic Acids Res. 2018; 46 (W1):W408-W416. [View at Publisher] [DOI:10.1093/nar/gky377] [PubMed] [Google Scholar]
33. Varma PBS, Yesubabu A, Subrahmanyam K. Identify virtual ligand hits using consensus scoring approach for drug target S. Aureus Int J of Eng & Tech. 2018; 7 (2.7) 84-87. [View at Publisher] [DOI:10.14419/ijet.v7i2.7.10265]
34. Bowie JU, Luthy R, Eisenberg D. A method to identify protein sequences that fold into a known three-dimensional structure. Science. 1991; 253: 164-170. [View at Publisher] [DOI:10.1126/science.1853201] [PubMed]
35. Mustufa MMA, Chandra S, Wajid S. Homology modeling and molecular docking analysis of human RAC-alpha serine/threonine protein kinase. Int J Pharma Bio Sci. 2014; 5: 1033-1042.
36. Prajapat R, Jain S, Vaishnav MK, Sogani S. In Silico Characterization of Surface Glycoprotein (QHD43416) of Severe Acute Respiratory Syndrome-Coronavirus. Chinese J Med Res. 2020; 3(2): 32-36. [View at Publisher] [DOI:10.37515/cjmr.091X.3201] [Google Scholar]
37. Benkert P, Biasini M, Schwede T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics. 2011, 27: (3) 343-350. [View at Publisher] [DOI:10.1093/bioinformatics/btq662] [PubMed] [Google Scholar]
38. Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 1993;2(9):1511-1519. [View at Publisher] [DOI:10.1002/pro.5560020916] [PubMed] [Google Scholar]
39. Newman JA, Douangamath A, Yadzani S, Yosaatmadja Y, Aimon A, Brandão-Neto J, Dunnett L, Gorrie-Stone T, Skyner R, Fearon D, Schapira M, von Delft F, Gileadi O. Structure, mechanism and crystallographic fragment screening of the SARS-CoV-2 NSP13 helicase. Nat Commun. 2021; 12(1):4848. [View at Publisher] [DOI:10.1038/s41467-021-25166-6] [PubMed] [Google Scholar]
40. White MA, Lin W, Cheng X. Discovery of COVID-19 Inhibitors Targeting the SARS-CoV-2 Nsp13 Helicase. J Phys Chem Lett. 2020; 11(21):9144-9151. [View at Publisher] [DOI:10.1021/acs.jpclett.0c02421] [PubMed] [Google Scholar]
41. Wiederstein M, Sippl MJ. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007; 35: W407-W410. [View at Publisher] [DOI:10.1093/nar/gkm290] [PubMed] [Google Scholar]
42. Rekik I, Chaabene Z, Grubb CD, Drira N, Cheour F, Elleuch A. In silico characterization and molecular modeling of double-strand break repair protein MRE11 from Phoenix dactylifera v degletnour. Theor Biol Med Model. 2015; 12:23. [View at Publisher] [DOI:10.1186/s12976-015-0013-2] [PubMed] [Google Scholar]
43. Wiederstein M, Sippl MJ. Protein sequence randomization: Efficient estimation of protein stability using knowledge-based potentials. J Mol Biol. 2005; 345: 1199-1212. [View at Publisher] [DOI:10.1016/j.jmb.2004.11.012] [PubMed] [Google Scholar]
44. Benkert, P., Biasini, M., Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics. 2011; 27, 343-350. [View at Publisher] [DOI:10.1093/bioinformatics/btq662] [PubMed] [Google Scholar]
45. Studer G, Rempfer C, Waterhouse AM, Gumienny G, Haas J, Schwede T. QMEANDisCo - distance constraints applied on model quality estimation. Bioinformatics. 2020; 36: 1765-1771. [View at Publisher] [DOI:10.1093/bioinformatics/btz828] [PubMed] [Google Scholar]
46. Freidel MR, Armen RS. Mapping major SARS-CoV-2 drug targets and assessment of druggability using computational fragment screening: Identification of an allosteric small-molecule binding site on the Nsp13 helicase. PLoS One. 2021; 16(2): e0246181. [View at Publisher] [DOI:10.1371/journal.pone.0246181] [PubMed] [Google Scholar]
47. Vardhan S, Sahoo SK. Exploring the therapeutic nature of limonoids and triterpenoids against SARS-CoV-2 by targeting nsp13, nsp14, and nsp15 through molecular docking and dynamics simulations. J Trad Compl Med. 2022; 12(1): 44-54. [View at Publisher] [DOI:10.1016/j.jtcme.2021.12.002] [PubMed] [Google Scholar]
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
Enamad



This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.